|
|

|

|
| e-Pub |
Section: New Results
Parallel algorithms for adaptive molecular dynamics simulations
Participants : Dmitriy Marin, Stephane Redon.
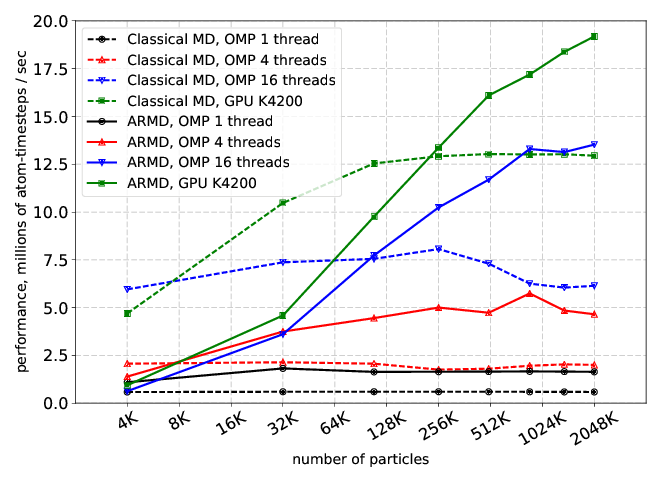
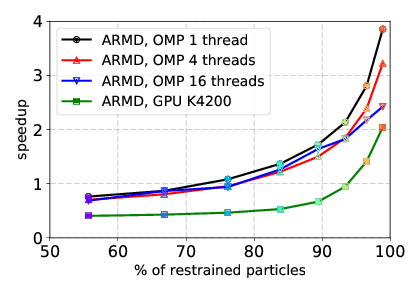
We worked on the development and improvement of a parallel implementation of the Adaptively Restrained Molecular Dynamics (ARMD) method in the LAMMPS molecular dynamics simulator. The parallelization was done in application to multi-core CPU and hybrid CPU/GPU systems thanks to the Kokkos package provided by LAMMPS. The ARMD can be used for decreasing computational complexity by restraining degrees of freedom for some particles in the simulated system [23], therefore allowing to gain speed-up by either decreasing precision or focusing on select subsystems. The developed parallel implementation allows us to run LAMMPS with an ARMD integrator on central processing units (CPU), graphics processing units (GPU), and many integrated core architecture (MIC). We developed a new algorithm for processing particles that switches their state from a restrained state to a full-dynamics state and vice versa. The new algorithm is modified for efficient usage of GPU and many-core CPUs (computations are performed on a computational device, communications between host and device are decreased). The results on performance and speed-up for ARMD in comparison with the non-modified LAMMPS for a standard Lennard–Jones liquid benchmark are shown in Figures 10 and 11. We showed that starting from some number of atoms in the system and from some percentage of restrained atoms, ARMD provides better performance over classical MD. The results are published in [14].